title
AlphaFold: is it worth the hype?
Google’s AI offshoot DeepMind has opened a new avenue “AlphaFold” to solve the 3D structure of the protein from its sequence and there is a lot of hype around that. Some even say that it is the greatest discovery in the field of artificial intelligence and machine learning which can potentially win the Noble Prize, while others feel it is just a black box for protein structure prediction.
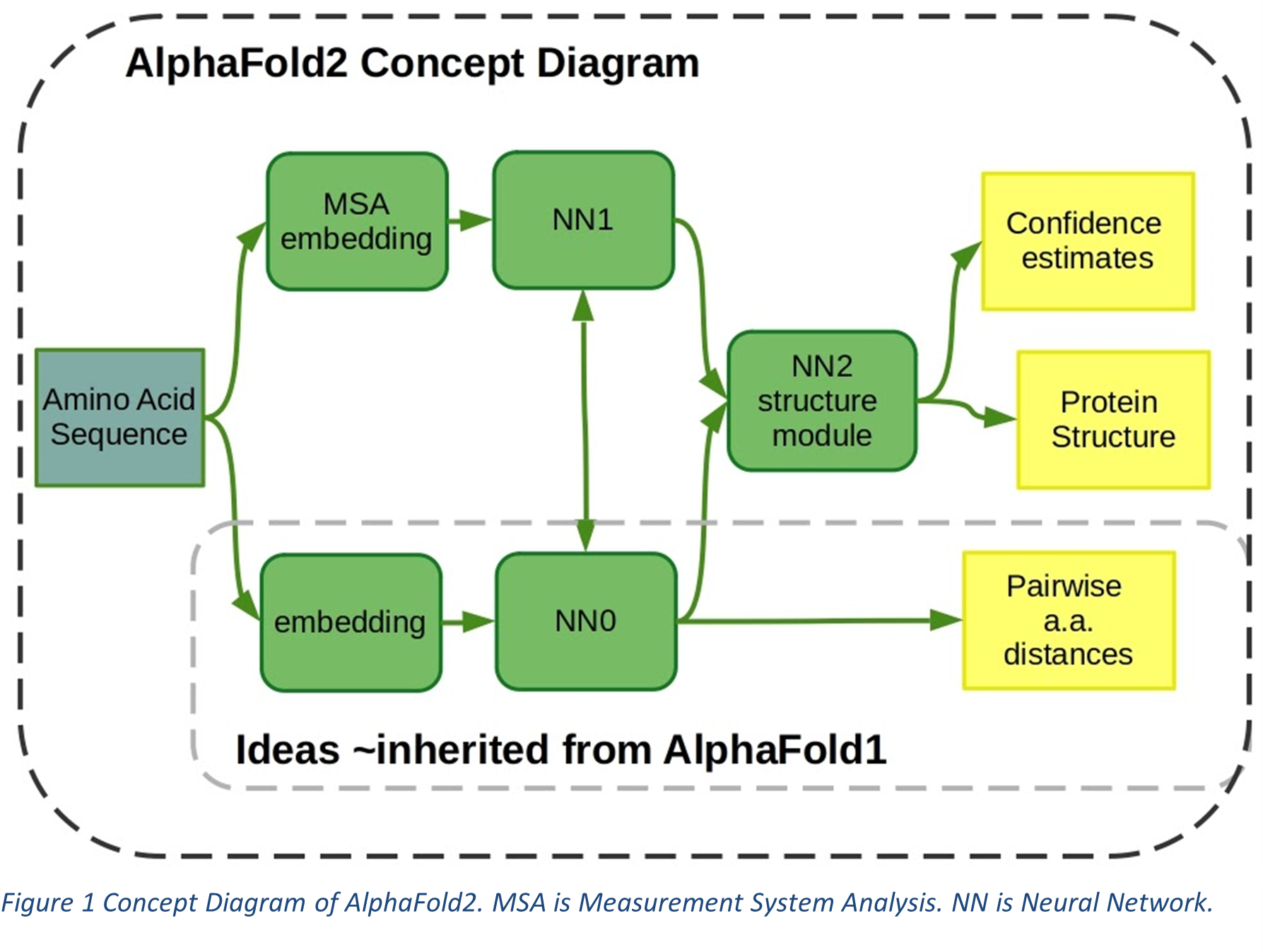
The problem of protein folding is a decade-old problem. People have waged bets on whether computers have the power to correctly predict the three-dimensional structure of protein since the 1960s. Ever since there has been great progress in the field of Artificial Intelligence (AI) and Machine Learning (ML) and it would not be an exaggeration to say that the release of AlphaFold by DeepMind has been perceived as one of the biggest scientific breakthroughs in recent times. Forbes called it “the most important achievement in AI-ever”. The new version of AlphaFold, AlphaFold2, is an improvisation on the previous AlphaFold (submitted in CASP13) which had a two-step process of training a neural network followed by steepest descent optimization. AlphaFold solves the protein folding problem using attention-based neural network system (see Figure 1). AlphaFold has been able to predict protein structure with an accuracy matching that of experimental methods. It uses approximately 16 TPUv3s (which is 128 TPUv3 cores or roughly equivalent to ~100-200 GPUs) that run over a few weeks.
It has been trained on the available protein structures in Protein Data Bank. It creates a force field by taking into consideration the distribution of C-β distances between pairs of residues. In addition, AlphaFold also uses Multiple Sequence Alignment from experimentally determined protein structures. This means that it is heavily dependent on experimental methods and cannot predict the 3-dimensional structure of protein only from its sequence. In addition, if there is a protein with a new fold that has not been discovered yet, then AlphaFold cannot predict the structure correctly.
The AlphaFold network received the highest score in protein structure prediction at the CASP14 (Critical Assessment of protein Structure Prediction) competition. Two-thirds of the predictions from AlphaFold were topologically correct, but it could not tell which were they, until, they were compared to experimental results. About 36% of the predictions were so precise that they can detail atomic features like the active site of an enzyme, which has implications for drug discovery.However, atomic positions had an average error of 1.6 Å, which is the scale of a bond length.
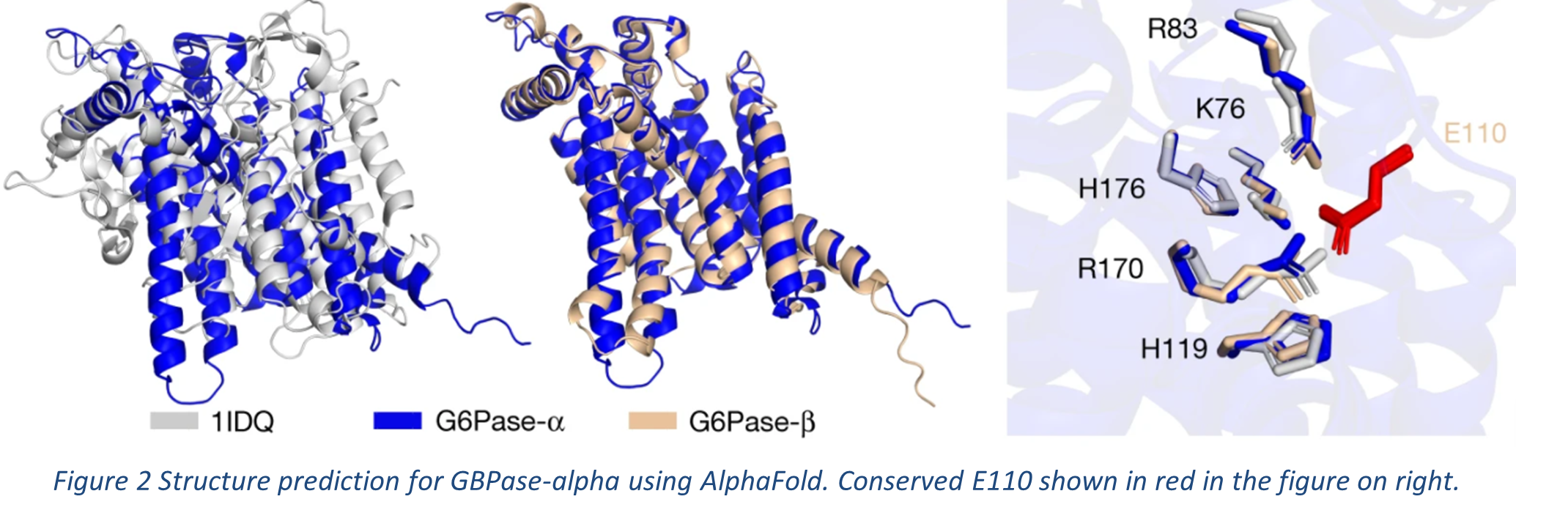
AlphaFold has also been used in predicting active sites in proteins. Glucose-6-Phosphatase (G6Pase-α) is an important enzyme that maintains blood sugar levels; it is membrane-bound and catalyses the final step in glucose synthesis. AlphaFold has predicted a high-confidence structure: a nine-helix topology and its active site. A conserved glutamate (E110), also present in G6Pase-β, was predicted opposite the G6Pase-α binding pocket. It faces the residues common with chloroperoxidase (PDB ID: 1IDQ), which is structurally similar to G6Pase-α (see Figure 2). The glutamate could stabilize the binding pocket in a closed conformation by forming salt bridges. It is also involved in gating mechanism.
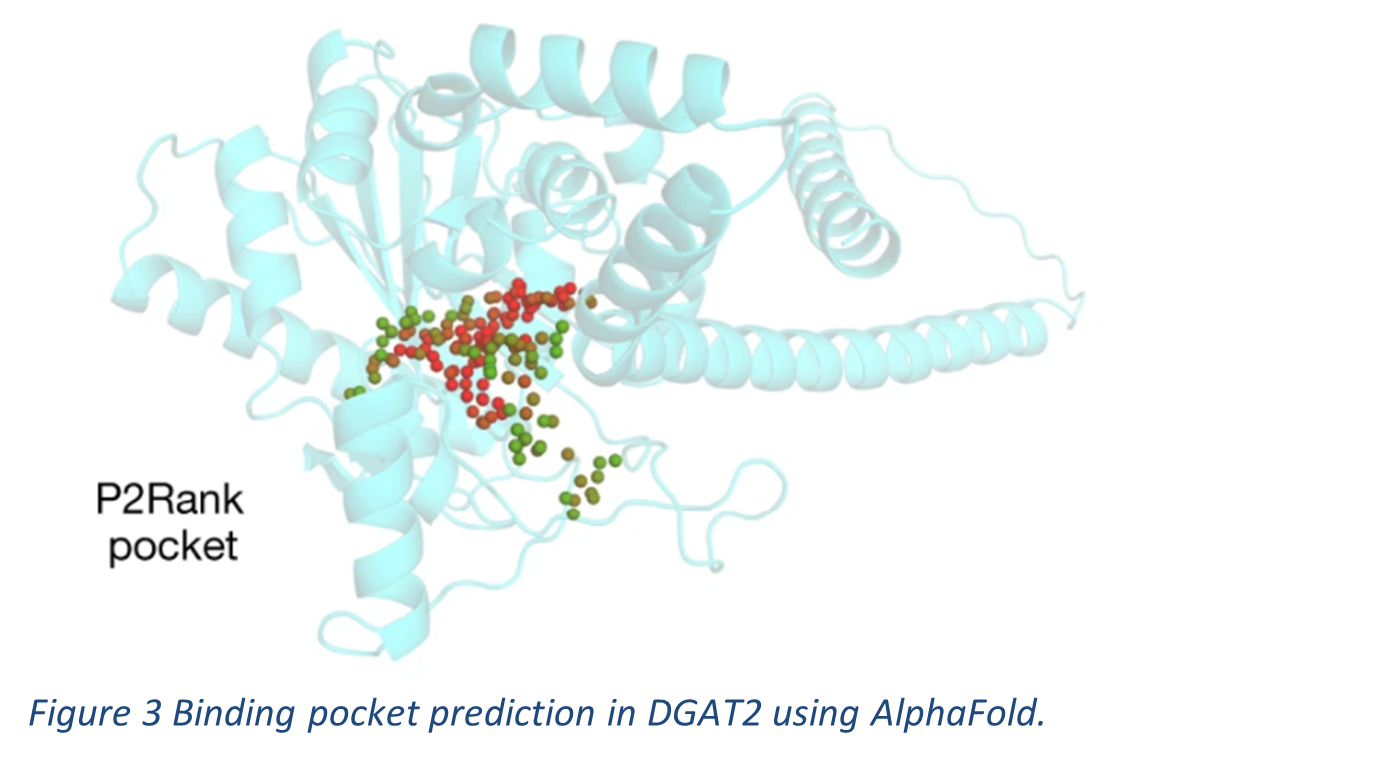
Diacylglycerol O-acyltransferase 2 (DGAT2) is another enzyme whose structure was predicted using AlphaFold (see Figure 3). It is an essential acetyltransferase that catalyses the final acyl addition in the process of storing metabolic energy as fat in adipose tissue. Inhibition of DGAT2 has been shown to improve liver function in animal models with liver disease. The high-confidence structure that was predicted showed a binding pocket where the inhibitor PF-06424439 was docked. The binding pocket residues: Glu243 and His163 – were analogous to the catalytic site residues in another DGAT, DGAT1. These residues are strong contenders for catalytic activity as they are conserved.
In spite of its modelling predictions, there are major drawbacks to AlphaFold. AlphaFold predictions have not been in agreement with structures obtained from nuclear magnetic resonance spectroscopy. Also, in some proteins, there is distortion in protein shapes based on their interaction with each other such as proteins with multiple functional units with linker elements. In such cases, AlphaFold does not give an accurate prediction. The newly proposed AlphaFoldMultimer is said to overcome the limitations of AlphaFold in predicting biological assemblies. Similarly, the prediction of novel enzymes by a tool like AlphaFold still has miles to go. Enzyme prediction involves the interplay between structure, binding, and catalysis. The study of enzyme-substrate interaction and dynamics is not possible by the current version of AlphaFold as it cannot predict cofactors, ions, and ligands such as heme, Zn2+, and ATP/ADP respectively.
A new tool, AlphaFill, has been proposed that claims to overcome some of the limitations of AlphaFold concerning missing ligands and cofactors. However, it also does not include glycosylation and phosphorylation. The AlphaFill models are said to be not suitable for protein-ligand interactions’ calculations. Thus, computational tools like AlphaFold are not directly suitable in enzyme engineering as of now. AlphaFold has limited value for modelling the effects of individual mutations which restricts the direct application of AlphaFold in in-silico based enzyme engineering process. In a recent Nature publication, the modelling of single point mutations by AlphaFold have been found inadequate to predict protein misfolding. However, if a crystal structure is not available, the model designed by AlphaFold can be a good starting point for docking of the ligand and further molecular dynamics simulations.
AlphaFold is a useful computational tool for protein structure prediction. However, is it worth the hype that has been created around it? It has a lot of limitations as described in this article. It is in no way a replacement for experimental techniques like NMR, X-ray crystallography, and cryo-electron microscopy. Moreover, it is not directly useful in real life problems like enzyme engineering and drug discovery in its current version. New tools or further improvisation in existing tools might help in mitigating the limitations of AlphaFold in these fields.
References