author_name
Aqza Elza John
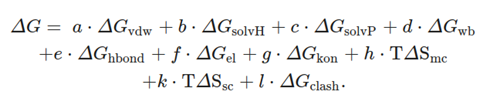
Enzymes are proteins that function as biological catalysts to accelerate chemical reactions. The linear sequence of amino acids formed by the peptide bonds make the primary structure of the protein. This primary structure then folds in a certain manner based on the interaction between the parts of the protein chain resulting in the secondary structure. It is classified as alpha helix and beta sheets which is formed by hydrogen bond between N-H group and C=O group in the backbone of protein chain.
Individual amino acids in the primary sequence interact with one another to form secondary structures such as helices and sheets while individual amino acids from distant parts of the primary sequence intermingle via charge-charge, hydrophobic, disulfide, or other interactions to form tertiary structure. Hydrophobic collapse is one of the main events necessary for reaching a protein's stable and functional conformation. The formation of these bonds and interactions serve to change the shape of the overall protein.
Figure 1: Representation of alpha helix, beta sheet, hydrophobic interaction, and disulfide bond
Protein quaternary structure is the arrangement of several protein chains or subunits into a closely packed arrangement. The subunits are held together by hydrogen bonds, van der Waals interactions, salt bridges, covalent disulfide linkages etc. Protein folding is integral to proper functionality within biological systems. When a protein is folded correctly, these interactions create a stable and functional structure. Thus, Conformational stability are the various forces that help to keep a protein folded in the right way. Proteins perform extremely specific functions which are dependent on their structure. Proteins that do not fold correctly are non- functional and contribute nothing to a biological system.
The stability of a protein is an important factor that determines its structure, function, and overall effectiveness. Proteins can be unstable due to a variety of factors, including mutations, changes in temperature, pH, and the presence of other molecules. If a protein loses its stability, it can denature, meaning that it loses its native structure and can no longer perform its biological functions. Understanding protein stability is important for the development or the design of new proteins with specific functions.
Structure-Stability-Function Relationship
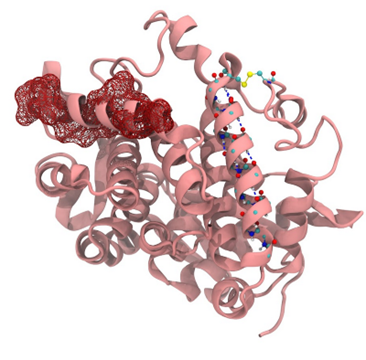
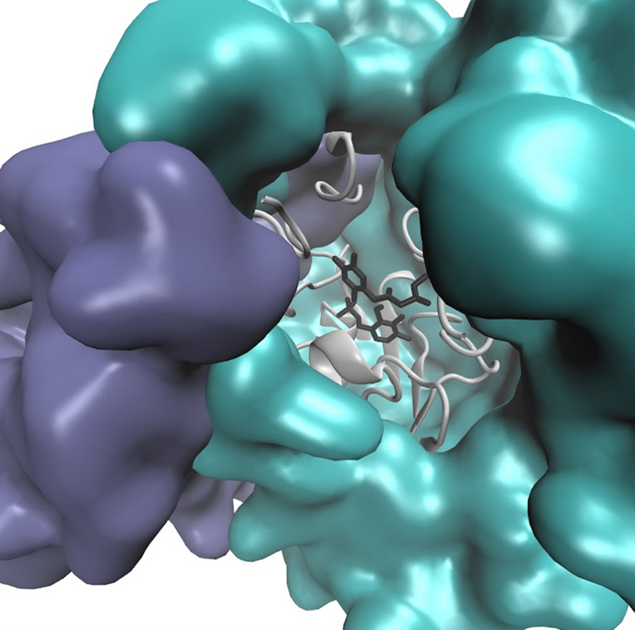
The part of the enzyme that directly binds to a substrate which is crucial for the biocatalytic reactions and lead to product formation is facilitated by the various amino acids in the active site. The active site of an enzyme is an important region which is formed by folding of protein chain of enzyme or coming together of different protein subunits.
Figure 2: Representation of the active site
There are other parts in the structure of enzyme that facilitates the entry and exit of substrate and product respectively.
Importance of Stability Prediction in Enzyme Engineering
Enzyme engineering is the process of customizing biocatalysts (enzymes) with improved properties by altering their constituting sequences of amino acids (called as mutations). If mutations are introduced without thorough understanding of the interactions or proper analysis, it can affect the protein structure, causing a destabilization that may compromise the protein activity. When an amino acid in Protein wildtype ‘W’ is mutated to variant ‘M’, the native interactions may be disrupted or fortified. The resulting change in stability can be determined by calculating ΔΔG, the thermodynamic stability of enzyme. Experimentally determination of the ΔΔG of each mutation is very time consuming and expensive.
Tools to predict the effect of mutations on the stability
There are various computational tools that can predict or calculate the impact of mutations on the stability of enzyme, whether it is destabilizing or stabilizing.
where ΔΔGMW represents the folding free energy change due to a mutation; ΔGW and ΔGM are folding free energy of the wildtype protein and the mutant, respectively.
Below mentioned are few out of many tools used in the stability check of Mutated enzymes.
They are Foldx, Rosetta, PoPMuSiC, CUPSAT, I –Mutant, mCSM, PremPS and Thermonet
FoldX, Rosetta and Thermonet are stand- alone tool whereas PoPMuSiC, CUPSAT, I-Mutant, mCSM and PremPS are web servers.
FoldX is an empirical force field- method. It calculates the effect of single point mutations through linear combination of empirical free energy terms, including electrostatic interaction, van der Waals forces, hydrogen bond, entropy contribution, solvation energy. The Full FoldX Suite is freely available to Academic and Non-Profit Research Institutions for research purposes only.
The following linear combination of empirical terms is used to calculate free energy (in kcal/mol) by FoldX
Rosetta is a method based on structural modelling that computes the difference in energy between the simulated wild-type and the mutated structures. Rosetta is available to all non-commercial users for free.
https://www.rosettacommons.org/software/
AI/ML based tools
Thermonet is a deep 3D-convolutional neural network designed for structure-based prediction of the ΔΔG values. Input protein structures are treated as if they were multi-channel 3D images, therefore by using multi-channel voxel grids based on biophysical properties derived from raw atom coordinates.
https://github.com/gersteinlab/ThermoNet
I-Mutant and CUPSAT is AI based web server where the model is trained based on data derived from ProTherm. The input files in I-Mutant are PDB ID, the chain id, the position of protein to be mutated and the residue to be mutated with. The pH and temperature can also be specified. I-Mutant and I-Mutant-Seq are the structure-based and the sequence version respectively of the I-Mutant method. It is an SVM based method whereas CUPSAT uses amino acid-atom potentials and torsion angle distribution to assess the amino acid environment of the mutation site. The input requires the protein structure. The output consists of information about mutation site, its structural features (solvent accessibility, secondary structure and torsion angles), and comprehensive information about changes in protein stability for 19 possible substitutions of a specific amino acid mutation.
https://folding.biofold.org/i-mutant/i-mutant2.0.html
PremPS is a random forest regression- based method that uses evolutionary and structure-based features to predict the free energy change. It has been trained on both stabilizing and destabilizing mutations so that the prediction will not be biased.
https://lilab.jysw.suda.edu.cn/research/PremPS/
PoPMuSiC is an energy function-based method providing a linear combination of 13 statistical potentials, two volume-dependent terms of the wild-type and mutant amino acids, and an independent term. The coefficients depend on the solvent accessibility of the mutated residue, based on a sigmoid function whose parameters are optimized through a neural network.
Most of these tools can predict only a single point mutation at a time.
The prediction of enzyme stability enables enzyme engineers to rapidly design enzyme variants with improved stability. This is particularly important in industrial biocatalysts, where enzymes are used to catalyse reactions on a large scale. Stable enzymes can be used for longer periods of time without losing activity with increased efficiency and turnover. Furthermore, predicting enzyme stability can aid in the design of new enzymes with improved properties.
In Quantumzyme, we apply this technology to predict the stability of enzymes during the engineering process to improve the functionality and to make the enzymes industry ready.
References