author_name
Shreya Shroff

Enzyme/Protein engineering is the field of scientific research and development that focuses on modifying and designing enzymes. Enzyme engineering involves the manipulation of enzymes at the molecular level by means of chemical or genetic techniques for various applications such as identification of structural and functional relationships, enhancing the enzyme activity, broadening the enzyme specificity, solvent tolerance, and thermostability. Several enzyme engineering approaches have been successfully applied to industrially important enzymes for the synthesis of stereoselective and regioselective molecules.
Site-directed mutagenesis is a powerful technique used in enzyme engineering to introduce specific mutations at desired hotspots in the DNA sequence encoding an enzyme. This technique allows researchers to modify the amino acid sequence of the enzyme, which can lead to improved enzyme properties and functionalities.
Methods of site-directed mutagenesis
1. Overlap extension PCR
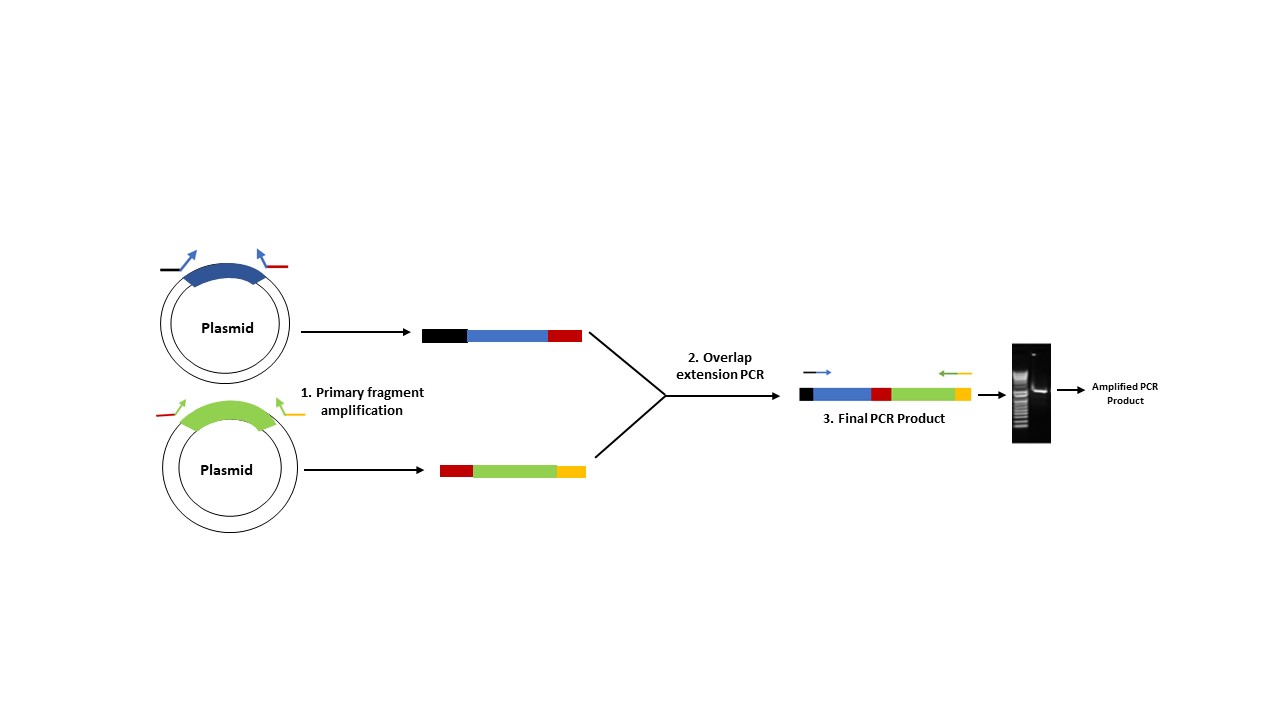
Overlap extension PCR (Polymerase Chain Reaction) is preferred when the experimental aim is to create chimeric DNA sequence by fusing two or more DNA fragments and in insertion of specific DNA sequence such as promoter, tags, or regulatory elements. In this method, two PCR products with overlapping ends are generated separately, each containing a portion of the desired mutation called primary strands. The second step of PCR i.e., overlap extension does not require additional primers. It relies on the overlapping primary fragments generated in the step 1. These primary fragments are then mixed and subjected to a second round of PCR, which results in a full-length mutated product (Figure 1). This method can be used to introduce multiple mutations simultaneously. However, this method requires subcloning of the amplified PCR product.
Figure 1. Principle of Overlap extension PCR.
The agarose gel demonstrates the presence of an amplified product generated using overlap extension PCR. Lane 1 – DNA ladder, Lane 2 – Amplified DNA product
2. Nested PCR
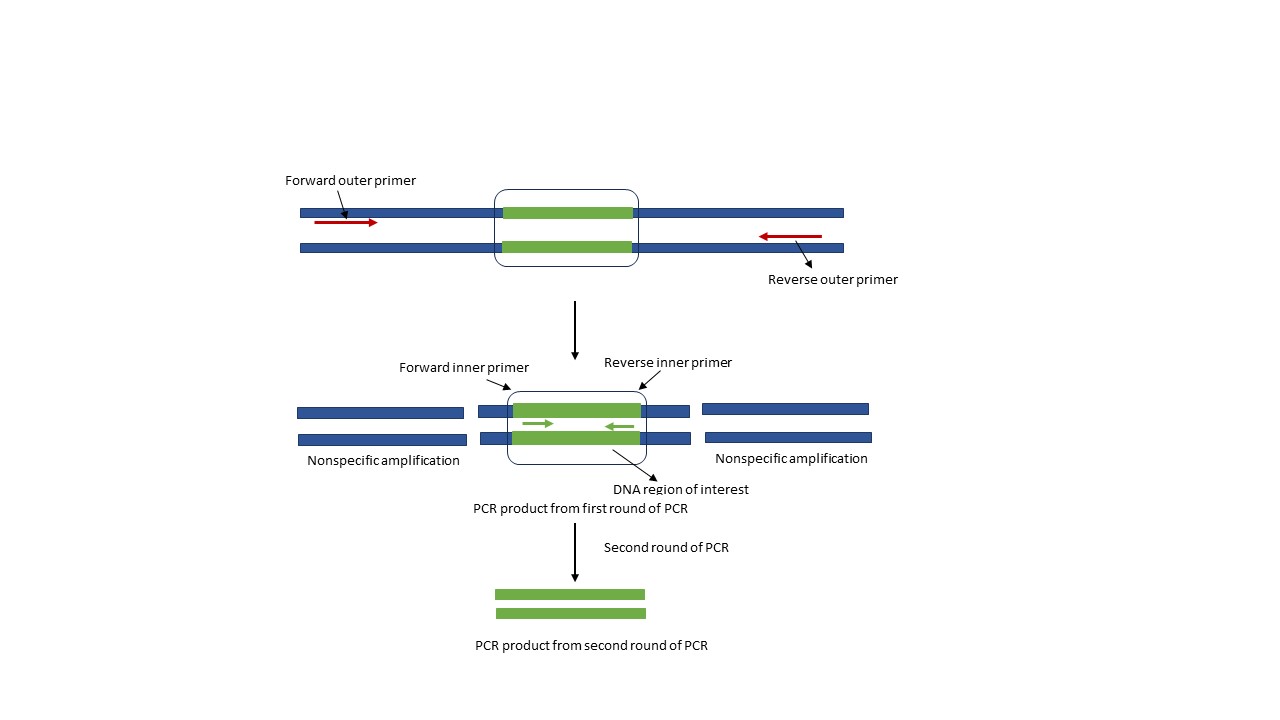
Nested PCR is a modification of the traditional PCR method to achieve higher specificity and sensitivity. This method is applicable when the target gene is present in very low level in the sample. The process of nested PCR involves two rounds of amplification using specific sets of primers. The first set of primers (outer primers) anneal to the region upstream to the second set of primers. These primers bind to the region outside of the target DNA. This results in amplification of entire template DNA with the region of interest as well as the regions. Even though, nonspecific amplification is observed in the first step of amplification, this is not carried in the second step. The second set of primers (inner/nested primers) are designed to bind specifically to the DNA region of our interest (Figure 2). It is very unlikely that these inner primers have binding sites in the unwanted PCR product, ensuring second step of PCR does not have any contamination from unwanted PCR products.
Figure 2. Principle of Nested PCR
3. CRISPR/Cas9-mediated site-directed mutagenesis
Site-saturation-directed mutagenesis libraries can be quickly constructed using a CRISPR/Cas9-mediated mutagenic method without the need for PCR. To eliminate the target nucleotides, Cas9 carrying single guide RNAs is used to double digest the plasmid DNA. The linearized plasmid's 5' ends are then excised by T5 exonuclease to produce homologous sections that are around 15 nucleotides long. The desired mutation is then introduced into the plasmid via base pairing in a brief dsDNA fragment of about 30–50 bp that cyclizes the plasmid. Following transformation, the host cells of Escherichia coli use a DNA repair process to fill in the gaps. This approach is very precise and effective. Site-directed mutagenesis can be successfully carried out on big plasmids, larger than 4.5 kb. This technique reveals the enormous potential for producing superior mutants.
4. PCR-based site-directed mutagenesis
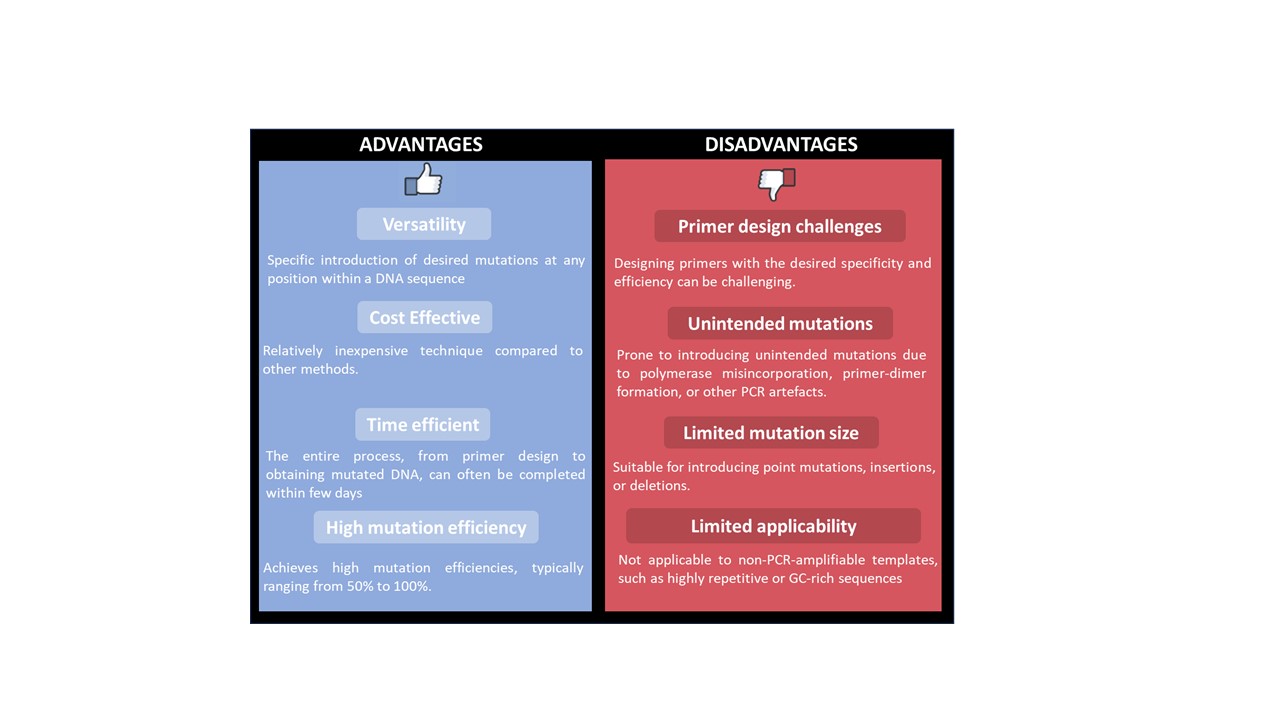
PCR based mutagenesis is widely used technique in molecular biology to introduce specific mutations into the DNA sequence. This allows the generation of variants with desired changes by altering the genetic information of the target gene. This technique relies on the principle of PCR. In this method, the designed primers incorporate the desired mutation followed by the amplification of the target DNA sequence. This technique has several advantages and disadvantages (Table 1).
Figure 3. Advantages and disadvantages of PCR based site directed mutagenesis
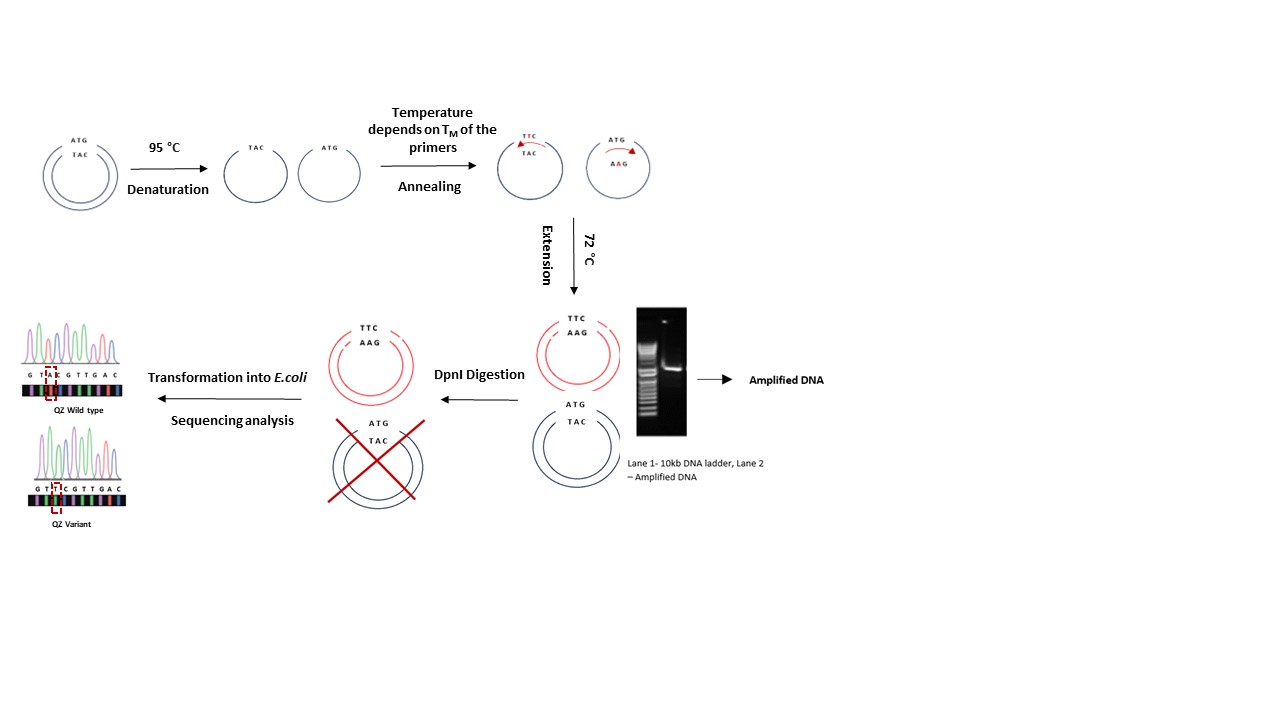
DNA with specific mutations can be synthesized using the mutagenic primers carrying desired mutation and amplifying the gene of interest using a PCR based protocol as shown in figure 4. After PCR, the parent template can be degraded using DpnI enzyme that cleaves the phosphodiester bond between the 5' phosphate and the 3' hydroxyl groups of adjacent nucleotides. Further, the amplified plasmid DNA (plasmid DNA with desired mutation) is transformed using a recA- strain such as DH5α/Top10. Plasmids are isolated from the resulting colonies with appropriate antibiotic resistance and sequenced to confirm the mutation (Figure 4)
Figure 4. Process of PCR based mutagenesis
Though, PCR based mutagenesis is simple and easy to perform there are few recommendations to be followed for successful enzyme engineering as listed below:
After the clone confirmation by nucleotide sequencing, the next step is to express the engineered protein in an expression host strain such as BL21(DE3). Further, the expressed protein will be isolated from the host and used in the biocatalytic reactions. The reactions are analysed by analytical methods such as TLC, HPLC, GC, etc.
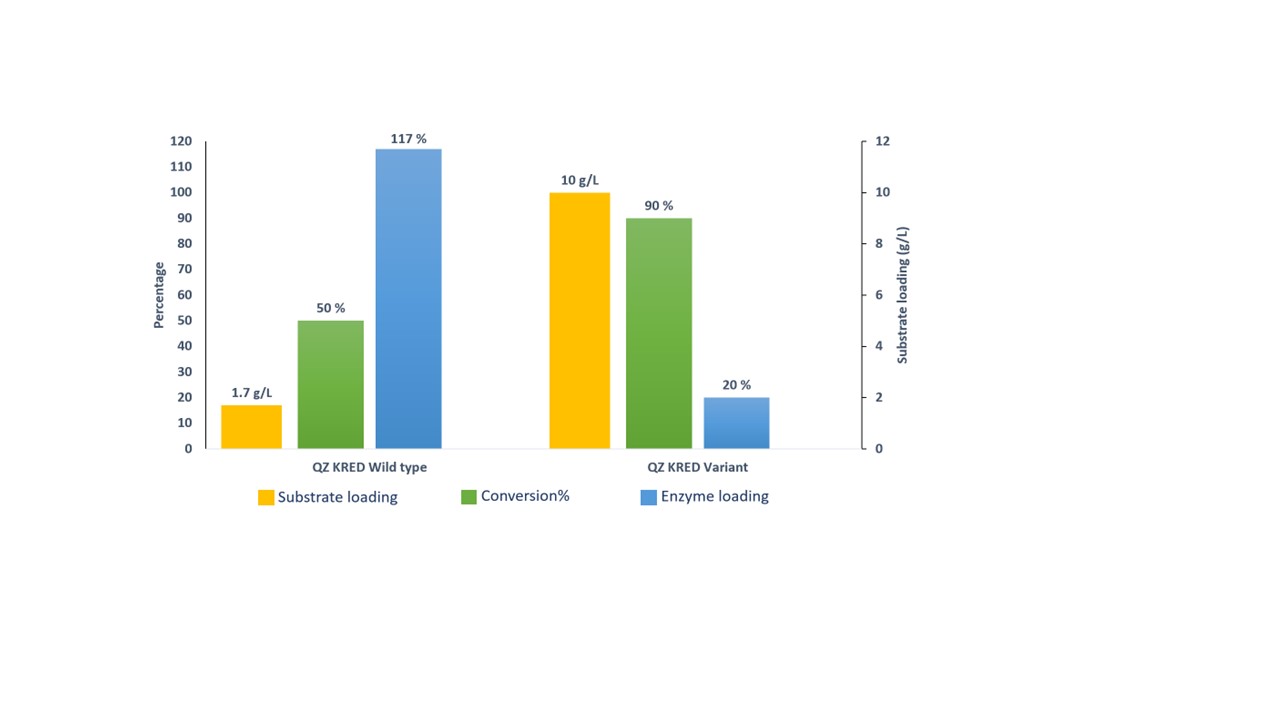
At Quantumzyme, PCR based mutagenesis has been applied in several instances to improve the conversion of substrate to product, to increase the substrate loading, and to establish the enantioselectivity etc. In our recent work, wild type gene encoding ketoreductase enzyme., has been engineered by insilico studies using Qzyme WorkbenchTM and were successfully produced invitro using PCR based site directed mutagenesis. This engineered variant has been developed with the target of improving bioconversion of substrate to product and substrate loading. More than 50% increase in the product formation was observed along with 5-fold increase in substrate loading based on the analysis done by RP-HPLC (Figure 5).
Figure 5. Comparision of QZ KRED wild type and QZ KRED variant
Overall, enzyme engineering using site-directed mutagenesis is a versatile tool that can be employed to fine-tune enzymes for specific biocatalytic applications. By introducing targeted changes in amino acid sequences, it is possible to improve the enzyme properties such as performance, stability, specificity, and compatibility with various reaction conditions, thus contributing to the advancement of sustainable and environment friendly industrial processes.
Reference: