author_designation
Research Assistant
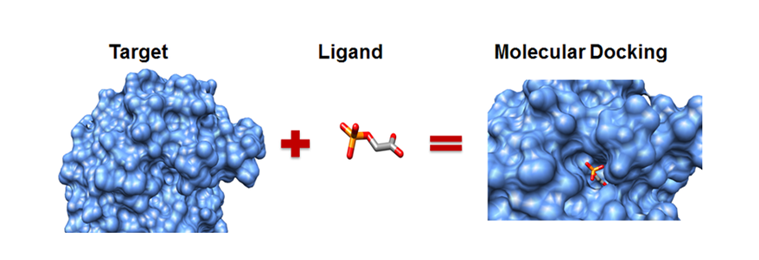
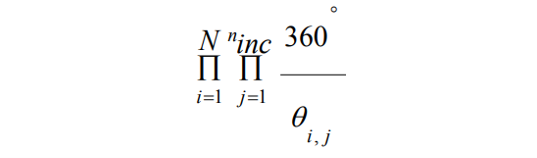
It is very well known that enzymes can catalyze chemical reactions in an organism, helping it sustain itself in a particular environment or to react to a particular kind of stress. This inherent capacity of enzymes to facilitate a chemical reaction, for example, oxidation, reduction, transamination, and many more, has been considered as an advantage in many chemical industries. Over the past decade, these enzymes have found extensive use in these chemical industries, especially to produce pharmaceuticals. While the primary focus of this article is to talk about different algorithms in molecular docking, it is important to first know the purpose of docking.
Molecular Docking: A Glimpse into Molecular Interactions
Molecular docking is a powerful technique that allows scientists to delve deep into the interactions between two molecular entities. At its core, molecular docking facilitates the understanding of interaction energy, which, in turn, enables the prediction of how two molecules will behave when placed in close proximity. This technique primarily revolves around two key players: the protein, known as the receptor, and the ligand. Molecular docking algorithm adeptly calculates the binding energy between the ligand and the receptor's binding site. The success of this endeavor is dependent upon the utilization of specialized algorithms and scoring functions, which enable researchers to assess the suitability of efficient enzymes in enzyme discovery. Here, we shall explore various algorithmic approaches and diverse scoring functions.
General steps involved in molecular docking
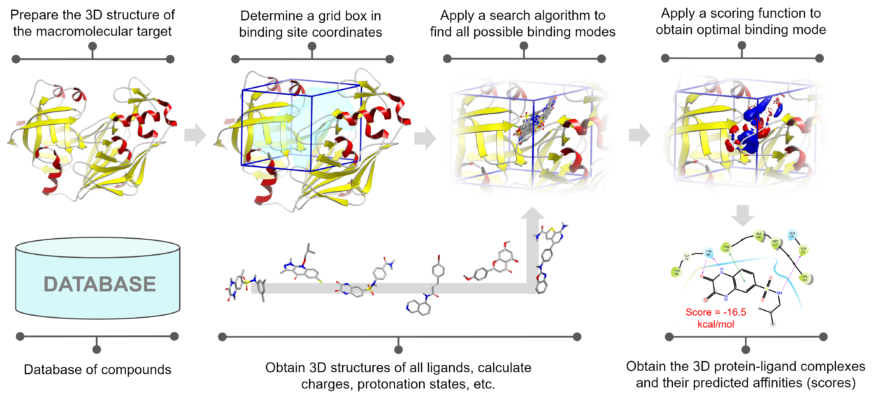
There are a series of steps that must be followed in the docking process. The most important step is the preparation of protein and ligand molecules. In this step usually, the polar Hydrogen is added, water and other unimportant molecules are removed, and charge is given to the residues in the protein and ligand. Once the protein and the ligand files are prepared, a grid box is set. This grid box is a reference area that says the ligand must bind within the given dimension. Once this is done the ligand can be docked into the protein.
Diverse Search Algorithms for Molecular Docking
Molecular docking employs various search algorithms, each with its own strengths and limitations:
1. Systematic Search Algorithms: These meticulously explore the conformational space by making incremental changes to structural parameters. It tries to explore all the degrees of freedom in a molecule such as bond rotations and angles and size of increment. However, they grapple with the challenge of combinatorial explosion due to the multitude of possible molecular conformations. The number of possible molecular conformations is given by,
Where N represents the number of rotatable bonds, ninc is the number of increments and θi,j represents the incremental angle j for bond i.
2. Exhaustive Search Algorithms: These methods systematically rotate all possible rotatable bonds within predefined intervals. Algorithms like GLIDE employ heuristics to narrow down the search space, making it more manageable.
3. Fragment-Based Algorithms: Fragmentation methods construct ligand conformations by docking individual fragments into the binding site, either one at a time or all at once. Ligand conformations are obtained by docking fragments in the binding site one at a time and incrementally growing them or by docking all fragments into the binding site and linking them covalently. The ligand is divided into rigid and flexible. The rigid part of the ligand is first docked and then the flexible parts are added incrementally.
4. Stochastic or Random Search Methods: These techniques introduce random changes to ligands, evaluating them based on a predefined probability function. Genetic algorithms, Monte Carlo simulations, and Tabu search fall into this category, providing diverse strategies for exploring binding interactions. This strategy avoids trapping the final solution at a local energy minimum and increases the probability of finding a global minimum. This is because the algorithm is designed to analyze the energy landscape in a wider range.
Scoring Algorithms: Unveiling Molecular Affinities
Molecular docking employs various scoring algorithms to evaluate potential binding orientations. This is a method of evaluating the various conformations obtained from search algorithms.
1. Force-Field Based Scoring: These scoring functions utilize energy terms like van der Waals interactions, electrostatic forces, and bond stretching/bending/torsional forces. Force-field based methods offer detailed insights but can be computationally intensive.
where rij represents the distance between protein atom i and ligand atom j. Aij and Bij are the van der Waals parameters, and qi and qj are the atomic charges. Here, ε(rij) is the distance dependent dielectric constant representing the effect of solvent implicitly.
2. Empirical Scoring: Empirical scoring functions expedite binding affinity estimation by relying on experimentally observed values. They include terms for ionic interactions, hydrogen bonds, lipophilic contacts, and the number of rotatable bonds in the ligand.
3. Knowledge-Based Scoring: Derived from statistical observations of intermolecular contacts, these scoring functions aim to reproduce structural configurations. Knowledge-based scoring function is expected to identify the near-native binding poses and at the same time provide a score that is close (or proportional) to the experimentally derived value They offer a balance between accuracy and computational efficiency, making them valuable tools for molecular docking.
Δij = 0 or 1 depending upon whether values of i and j are more than 5Å or within 5Å. pij and p represent the interatomic and averaged interatomic interactions. k is the Boltzmann constant and T represents the Kelvin temperature.
Drawback and Limitations of Docking
The primary constraint on molecular docking arises from the insufficient confidence of scoring functions in providing precise binding energies. This is because certain factors related to intermolecular interactions, like entropy change and the solvation effect, are rarely anticipated with accuracy. Furthermore, several intermolecular interactions that have been shown to be significant are rarely considered in scoring methods. For example, it has been shown that halogen bonding and guanidine-arginine interactions contribute to the affinity of protein-ligand binding, although they are not considered.
Accurately handling the water molecules in the binding pocket during the docking process remains a persistent issue. This is a challenging task that requires significant attention soon. The inefficient scattering by smaller atoms causes the x-ray crystal structures to lack the coordinate information of hydrogen. Ignoring water molecules that may be functioning as a bridging molecule between the ligand and the receptor can be inaccurate when the precise location of hydrogen is unknown.
Rigid receptors are a major challenge in the field of docking. Depending on the ligand it binds to, a protein can take on a variety of conformations. Rigid receptor docking will correspond to a single receptor conformation, which frequently results in false negatives. The reason this occurs is that a protein, which is typically overlooked in docking, can be in constant motion between various conformational states with comparable energies.
In conclusion, molecular docking has firmly established itself as an indispensable tool to predict binding modes and prioritize hits. Molecular docking is a vital tool in in-silico research, utilizing diverse search algorithms and scoring functions to predict binding affinities and molecular conformations. Its advantages lie in its ability to provide insights into binding interactions. However, it also faces challenges due to the complex nature of molecules. This also includes the computational demands of certain algorithms and the simplifications made in scoring functions. Steady progress is being made in improving the molecular docking algorithms and software. Molecular docking provides remarkable insights into binding affinities and molecular complementarity when utilized judiciously. With continued research improving docking methodologies to overcome today's challenges, molecular docking will continue empowering everything from enzyme engineering to drug discovery for years to come. Its combination of speed, scalability and informative power position it at the forefront of in-silico binding analysis.