author_designation
Scientist

Proteins, the building blocks of life, are essential for all living organisms. They are made up of long chains of amino acids, and their 3D structure dictates how they function. But determining that structure has always been a challenge for scientists. Traditionally, scientists have relied on experimental methods like X-ray diffraction or NMR analysis, which can be time-consuming and expensive. Computational models, like Modeller and AlphaFold 3, are transforming how we predict protein structures without needing these complex techniques. So, which tool should you use for your research?
Let us dive in and explore the differences.
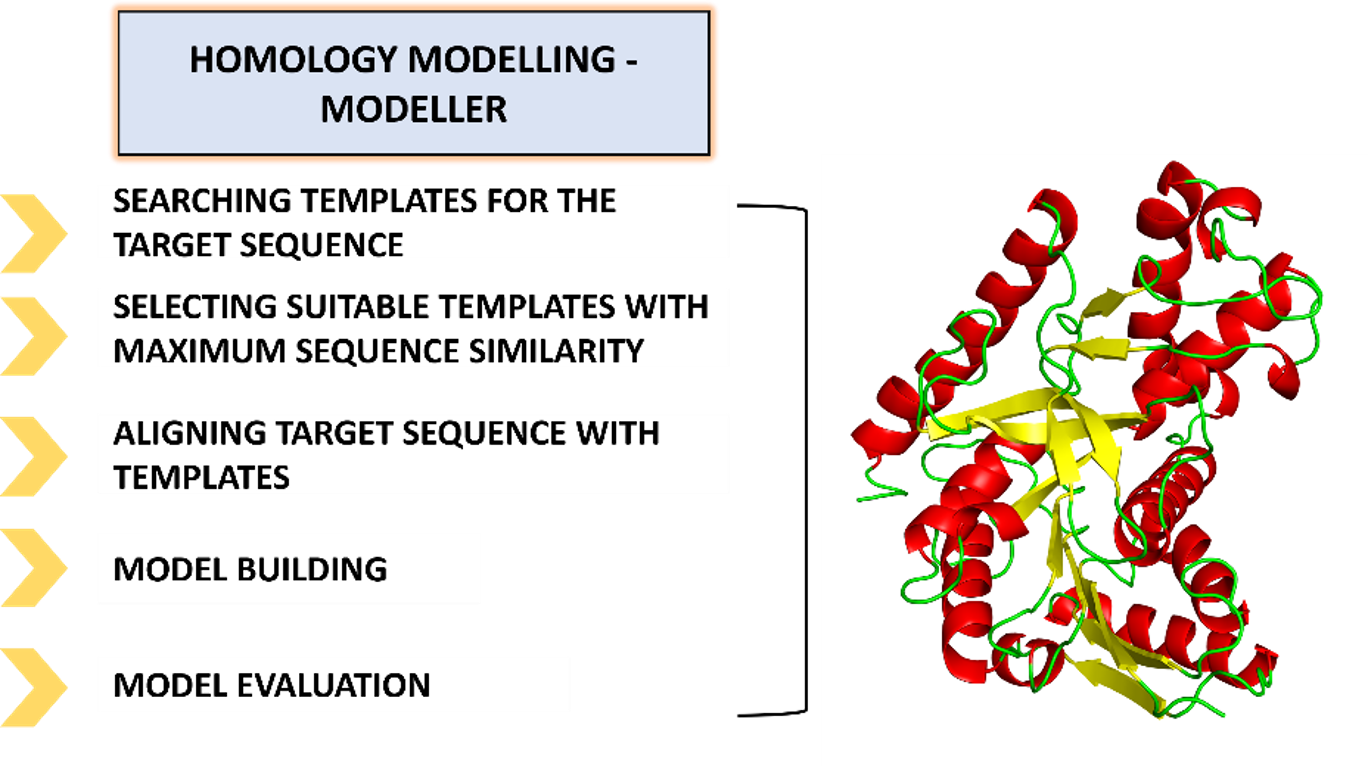
Modeller is one of the oldest and most trusted tools for predicting protein structures. It uses a technique called "homology modelling," which essentially compares your protein of interest to known protein structures. Think of it as looking for a family resemblance between proteins. Once Modeller has an idea of what your protein should look like, it tweaks and optimizes the model to get the best possible fit.
Using the data from an input target-template alignment, Modeller creates a set of homology-derived spatial restraints (HDSRs) that affect the atoms in the three-dimensional protein model. Homology-derived distance restraints (HDDRs) use sigma ("σ") values to specify how much conformational flexibility a model may have regarding its templates. MODELLER estimates σ values using a statistical method based on histograms. The optimization of the target structure is done by reducing the energy function through algorithm like the conjugate gradient method, which iteratively adjusts atomic positions to minimize the energy function, and simulated annealing, which involves heating and slowly cooling the system to escape local minima and find a more optimal global minimum. For regions that are not aligned correctly with the template sequence, such as loops, Modeller generates multiple conformations and selects the best conformation according to scoring functions like DOPE and GA341. The final models are further filtered and validated using experimental data and structural validation tools, ensuring the predictions are appropriate.
However, the catch is that Modeller heavily relies on how closely your protein matches existing structures. If your protein is novel or has few similar known proteins, Modeller’s predictions can be less accurate.
Figure 1: Basic steps of protein modelling using Modeller
AlphaFold 3, developed by DeepMind, it represents a significant leap in the field of protein prediction, using cutting-edge deep learning techniques to predict protein structures with near-experimental accuracy. Unlike Modeller, AlphaFold 3 doesn’t need a similar protein to compare against; it can predict completely new protein structures.
It processes input through multiple sequence alignments (MSA) and template data, capturing evolutionary and structural content. The neural network architecture includes transformers with multi-head self-attention mechanisms and the Evoformer module, which integrates MSAs and templates, enhancing the model’s understanding of protein structures. The structure module then directly analyses the three-dimensional coordinates of protein atoms and refines them iteratively, guided loss function like distogram loss and Frame Aligned Point Error (FAPE). AlphaFold 3 is trained on different protein datasets, that employ data augmentation and regularization techniques to enhance generalization and robustness. It provides high-confidence predictions with per-residue confidence scores (pLDDT) and predicted alignment error (PAE) scores. The model is validated benchmarking against experimental structures and blind prediction challenges like CASP.
What sets AlphaFold 3 apart is its ability to incorporate vast amounts of protein data into a neural network, learning from millions of protein structures to predict new ones. It is like having an expert who has seen every protein ever discovered and can make incredibly informed guesses about what your new protein looks like.
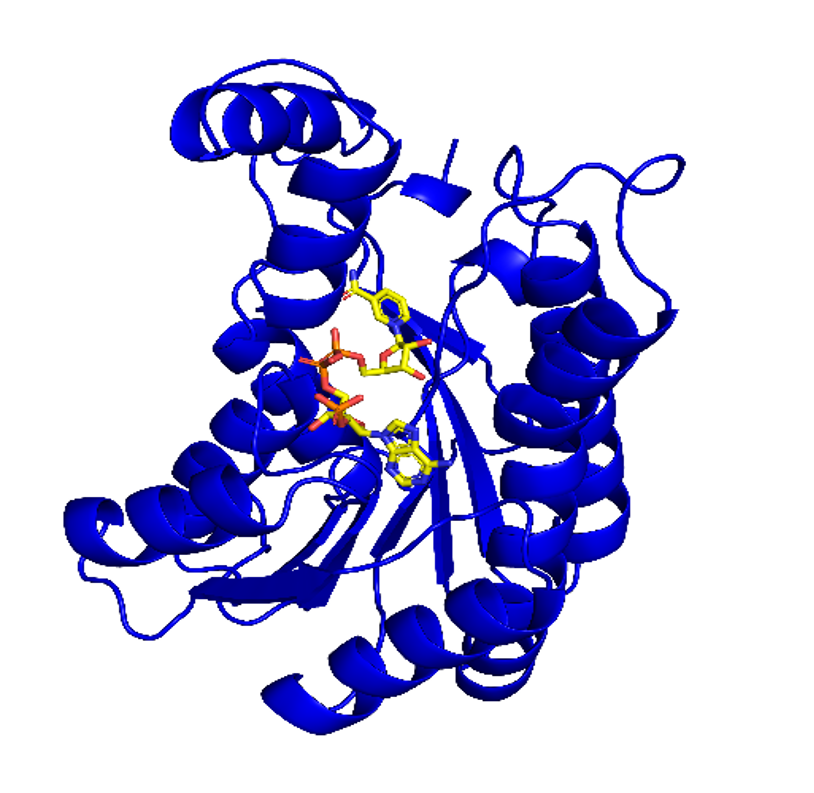
Figure 2: Dihydroanticaspin 7-dehydrogenase (pdb id: 5ITV) modelled with NADPH using AlphaFold 3.
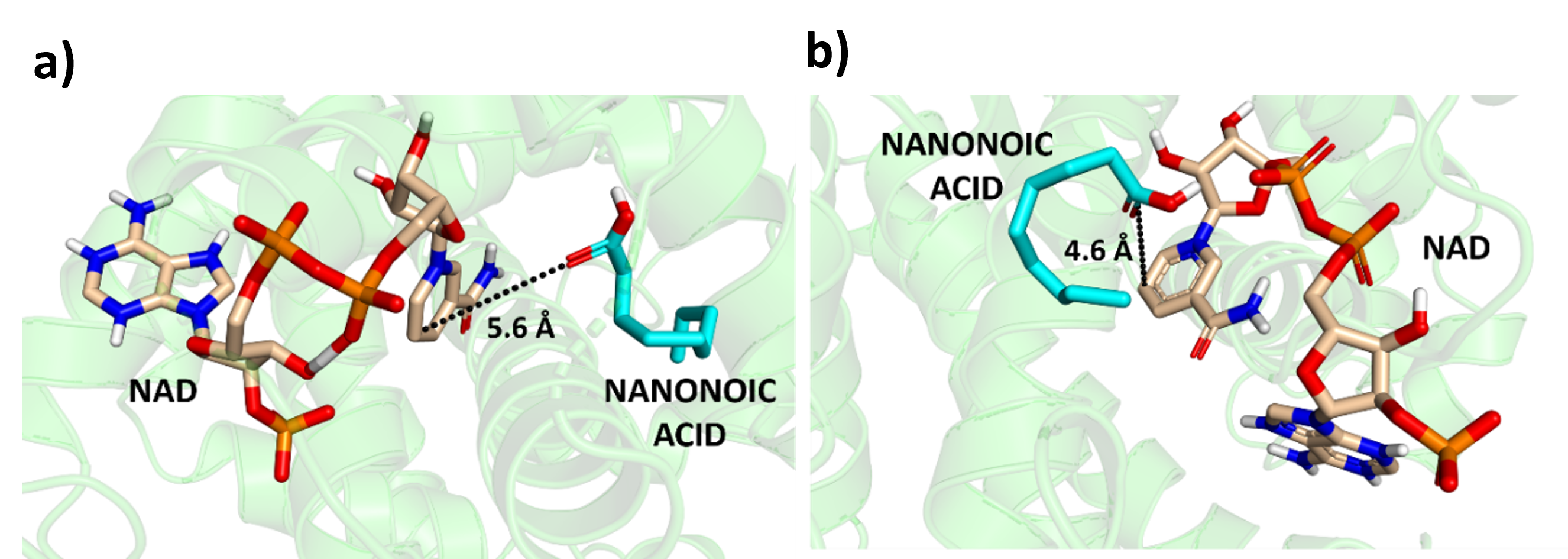
At Quantumzyme, we put both tools to the test by modelling the structure of a monooxygenase enzyme, a protein that did not have a known crystal structure. Then, we used molecular docking to study how well the models interact with a fatty acid substrate, nanonoic acid. Molecular dynamics (MD) simulations were then conducted to evaluate which model provided a more accurate representation of the sequence.
Figure 3: Best docking pose and least reactive distance of Protein modelled using a) Modeller and b) AlphaFold 3 respectively.
Binding energy of Protein Modelled with Modeller (kcal/mol) | Binding energy of Protein Modelled with AlphaFold3 (kcal/mol) |
-5.203 | -4.987 |
-5.166 | -4.656 |
-5.019 | -4.233 |
Table 1: Binding energy of the protein modelled with Modeller and AlphaFold 3 respectively.
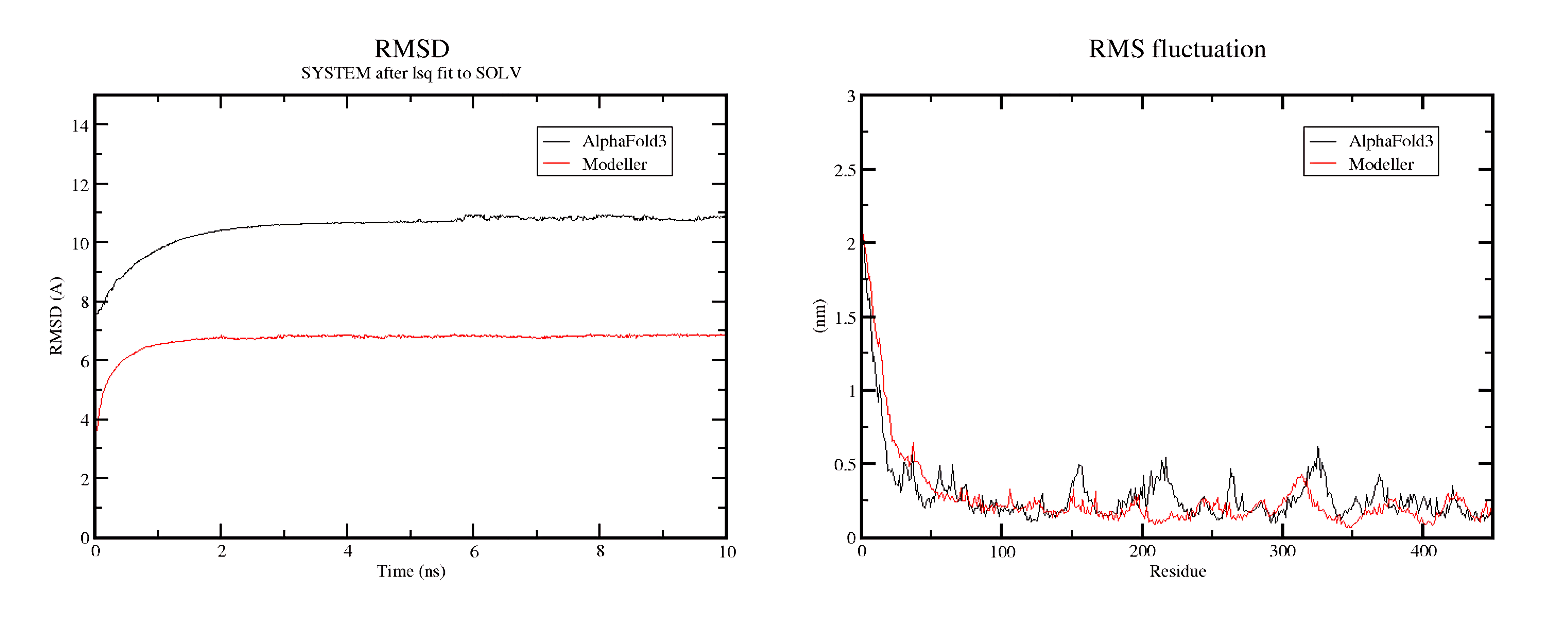
Figure 4: RMSD and RMS fluctuation values after MD simulation of 10 ns.
The results? Both tools produced reliable models, but Modeller’s structure showed slightly higher binding energy, meaning it formed a stronger interaction with the substrate. It also maintained a more stable structure throughout the molecular dynamics simulation, with a lower RMSD (a measure of how much the structure fluctuates). AlphaFold 3, while incredibly powerful, showed slightly more flexibility in the protein structure, which could be either a strength or a weakness depending on what you need. It may reflect a more dynamic model, sensitive to minor changes in the protein’s environment.
So, which tool should you use? It depends on your project. If you are working with a protein that has plenty of known relatives, Modeller offers more control and fine-tuning options. But if you are dealing with completely novel proteins, AlphaFold 3 is unbeatable in its ability to predict new protein structures.
Both tools are pushing the boundaries of what we can achieve in protein modelling, and together, they are helping scientists understand proteins in ways we never thought possible. Whether you are in drug design, bioengineering, or fundamental biological research, these tools will continue to drive breakthroughs.